
QE PWscf Band Structures
Published:
Content
- Theory
- 0. structural optimization, kpt convergence, ecut convergence, smearing convergence
- 1. scf
- 2. nscf
- 3. use
calculation = 'bands' - 4. bands.x post-processing
- 5. plotband.x
- 6. gnuplot
- 7. Python Plotting
- 8. Fatband
Band structure refers to the electronic band structure in solid state physics. In general, band structure is the k-E (or k-$\omega$) relation of the electrons distrbuting in crystaline, periodic systems. By analyzing band structure, electronic properties such as
- band gap and its type (direct or indirect), high symmetry points
- phase velocity ($v_{phase}=\frac{\omega}{k}$)
- group velocity ($v_{group}=\frac{d\omega}{dk}$)
- Fermi velocity
- effective mass of charge carriers
Theory
Updating…
The following is an example of the band structure calculation for 4H-SiC bulk crystal in 1x1x1 unit cell.
Example: 4H-SiC crystal, typical wide-bandgap semiconductor, usually for high-power electronics, substrate in semiconductor industry. 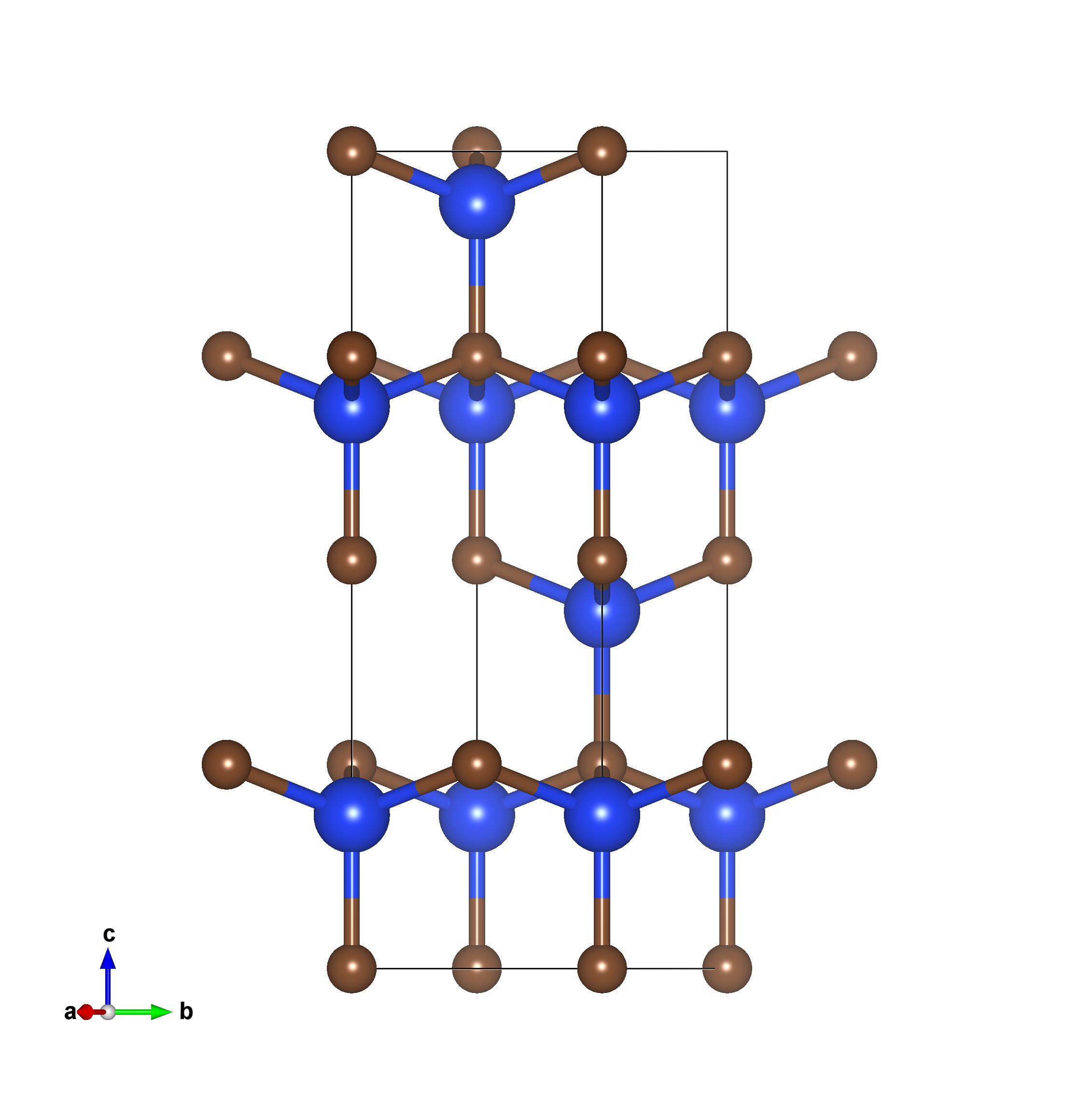
0. structural optimization, kpt convergence, ecut convergence, smearing convergence
Do before any calculation
1. scf
Dense enough K pt mesh
&CONTROL
title = '4H-SiC'
prefix = '4HSiC'
outdir = './calout'
pseudo_dir = '/home/yongnanli08/Desktop/4hsic/PP'
calculation = 'scf'
nstep = 1000
iprint = 1 ! write band information each iprint step
tprnfor = .true. ! calculation of force
tstress = .true.
restart_mode = 'from_scratch'
verbosity = 'high'
forc_conv_thr = 1e-5 ! no need if not relax vc-relax
etot_conv_thr = 1e-6 ! no need if not relax vc-relax
/
&SYSTEM
nat= 8
ntyp= 2
ibrav= 0
ecutwfc = 90.00
nosym = .true. ! no need, use for low-symmetric system (low-dimensional, etc)
force_symmorphic = .true. ! no need, force the symmetry group to be symmorphic, for yambo init
! =========== Smearing ===============
occupations = 'smearing'
smearing = 'mp'
degauss = 1e-3
/
&ELECTRONS
electron_maxstep = 500
mixing_mode = 'plain'
mixing_beta = 0.4
mixing_ndim = 12
conv_thr = 1.0d-11
diagonalization = 'david'
diago_thr_init = 5e-6 ! for charge density, 1e-5, can reduce if not converged
/
&IONS
ion_dynamics = 'bfgs' ! for structural optimization, relax & vc-relax
/
&CELL
cell_dynamics = 'bfgs' ! for structural optimization, relax & vc-relax
/
ATOMIC_SPECIES
Si 28.085 Si.pbe-n-rrkjus_psl.1.0.0.UPF
C 12.011 C.pbe-n-kjpaw_psl.1.0.0.UPF
CELL_PARAMETERS angstrom
3.099315860 -0.000000000 0.000000000
-1.549657930 2.684086271 0.000000000
0.000000000 0.000000000 10.114801588
ATOMIC_POSITIONS angstrom
Si 1.5496594817 0.8946945267 9.4877890025
Si -0.0000015518 1.7893917439 4.4303882085
Si 0.0000000000 -0.0000000000 6.9598048955
Si 0.0000000000 0.0000000000 1.9024041015
C 1.5496594817 0.8946945267 7.5821765481
C -0.0000015518 1.7893917439 2.5247757541
C 0.0000000000 0.0000000000 5.0603888110
C 0.0000000000 -0.0000000000 0.0029880170
K_POINTS automatic
8 8 8 0 0 0
2. nscf
actually no need (bands calculation is also nscf), the charge density has been determined in scf
-
calculation = 'scf'tocalculation = 'nscf' - k pt
8 8 8to16 16 16
3. use calculation = 'bands'
K_POINTS crystal_b
4 ! num_of_high_symmetry_points
0.0000000000 0.0000000000 0.0000000000 30 ! Gamma
0.5000000000 0.0000000000 0.0000000000 20 ! M
0.3333333333 0.3333333333 0.0000000000 30 ! K
0.0000000000 0.0000000000 0.0000000000 20 ! Gamma
The numbers following the coordinates are, the point interval between this high symm. point to the next one while doing interpolating. High symmetry path can check by upload bulk pwscf input file to seekpath. High symmetry point coordinates are in reduced form, in reduced first Brillouin zone!
Can also use Python script KptPath with K_POINTS crystal, same function with K_POINTS crystal_b.
4. bands.x post-processing
bands.x
&BANDS
prefix = ''
outdir = ''
filband = '_qebands.dat'
/
will get
- _bands.dat
- _bands.dat.gnu
- _bands.dat.rap
Run (I prefer to call this as a postprocessing opeartion, so with pp), can mpirun
bands.x < .ppbands.in > ppbands..out
5. plotband.x
don’t trust its output of high-symmetry point: , it is in cartesian coordinates, but we want high symm. point in crystal/reduced coordinates!
> plotband.x
Input file
> 4HSiCslabC_bands.dat
Reading 24 bands at 71 k-points
Range: -13.9960 7.1910eV Emin, Emax, [firstk, lastk]
> -6,8
high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 0.0000
high-symmetry point: 0.5000 0.2887 0.0000 x coordinate 0.5774
high-symmetry point: 0.3333 0.5774 0.0000 x coordinate 0.9107
high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 1.5773
output file (gnuplot/xmgr)
> gnuplot
bands in gnuplot/xmgr format written to file gnuplot
output file (ps)
> (press enter)
stopping ...
generate several gnuplot files, splited by high-symmetry points
6. gnuplot
gnuplot -p -e "plot 'gnuplot.1.1' w l, 'gnuplot.2.1' w l, 'gnuplot.3.1' w l, [0:1.5773] 3.8937 w l t 'Fermi level'"
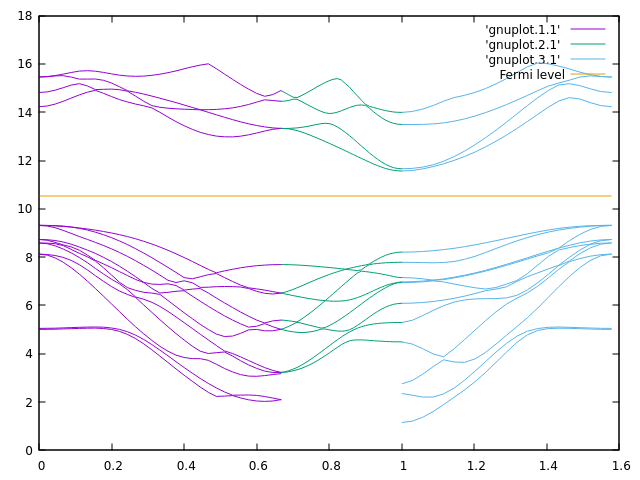
Obviously an indirect band gap is found, between Gamma point as VBM and K point as CBM.
BEAWARE: the use of plotbands.x may introduce some numerical error in post-processing, use
gnuplot -p -e "plot 'prefix_bands.gnu' w l, [0:1.5773] 3.8937 w l t 'Fermi level'"
is better, and then split the band by k point path by yourself (can see plotbands.x ‘s output to know the x coordinate of each high sym. point).
7. Python Plotting
use Bandstructure
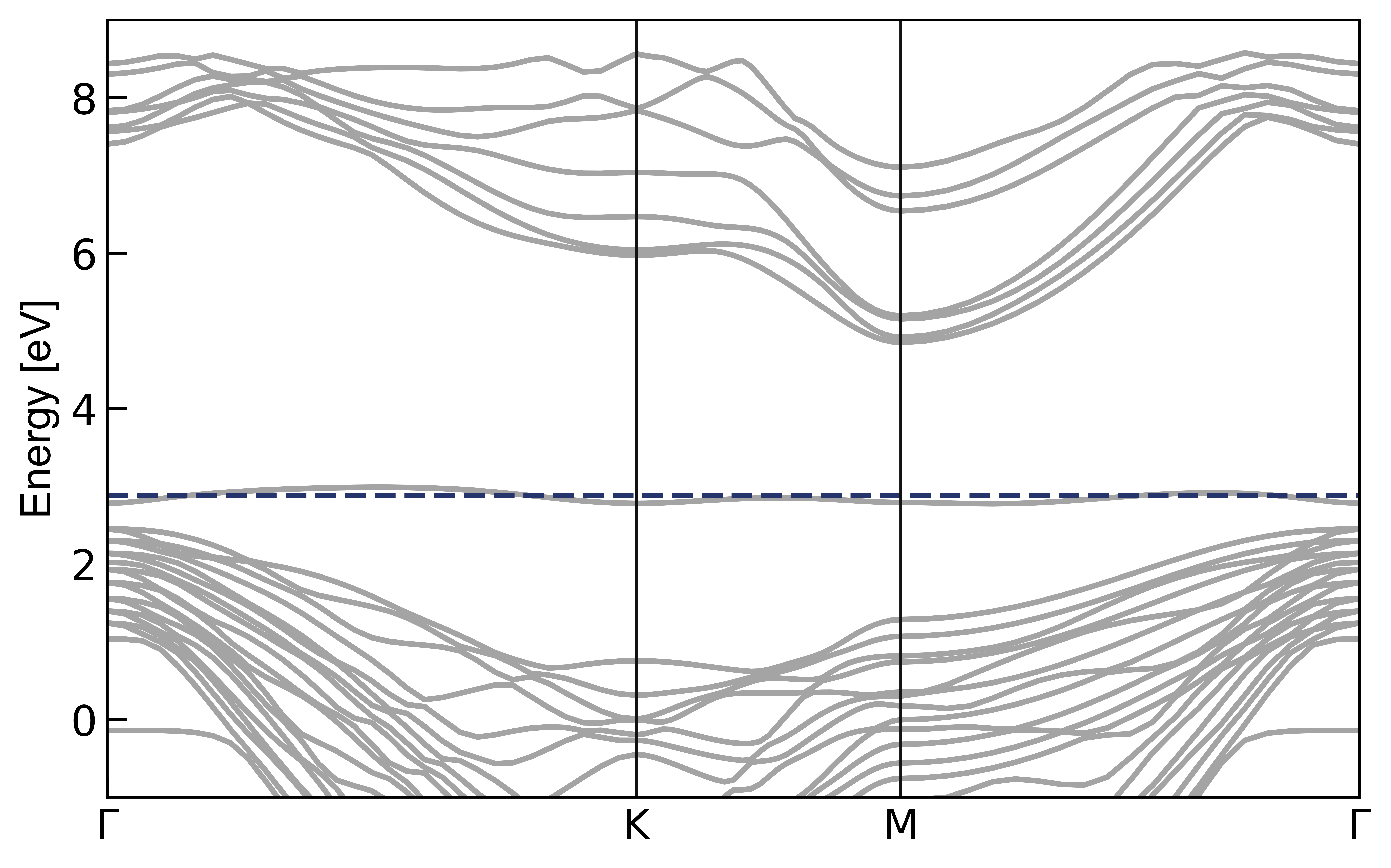
8. Fatband
Characteristics of band in our interested-area, and to see which of them are highly hybridized. This analysis called ‘Fatband analysis’. To do this, need to project the atomic orbitals to the Bloch wavefunction and then plot band structure. In simple words, to see the fractional contribution of atomic orbitals to the band structure.
After doing bands.x postprocessing, do another projwfc.x wavefunction projection:
projwfc.x
&projwfc
prefix = ''
outdir = './calout'
DeltaE = 0.01
filpdos = './PDOS/_pdos.dat'
filproj = './PROJ/_qebands.dat'
lsym = .false. ! symmetrized
lwrite_overlaps = .false. ! output of overlap matrix of atomic orbitals, in xml
kresolveddos = .false. ! k resolved DOS
tdosinboxes = .false. ! local DOS integrated in volumes
plotboxes = .false. ! Boxes in xsf format
/
Suggestion
Recommend to create a directory ./PROJ and /PDOS to store the output files, can do the similar thing in bands.x:
filband = './PROJ/_bands.dat'
and then run, can mpirun, faster
projwfc.x < .projwfc.in > projwfc..out
Be aware here no PDOS output, get output files
- _bands.dat.projwfc_up
change the name from
- _bands.dat.projwfc_up
to
- _bands.dat.proj
the purpose is to let plotband.x recognise the total part and projected parts of bands structures are generated.
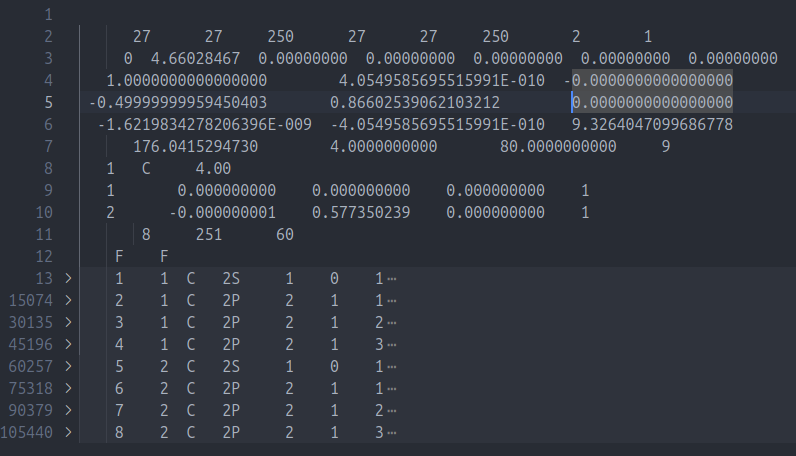
_bands.dat.proj
file look like this
27 27 250 27 27 250 2 1
0 4.66028467 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000
1.0000000000000000 4.0549585695515991E-010 -0.0000000000000000
-0.49999999959450403 0.86602539062103212 0.0000000000000000
-1.6219834278206396E-009 -4.0549585695515991E-010 9.3264047099686778
176.0415294730 4.0000000000 80.0000000000 9
1 C 4.00
1 0.000000000 0.000000000 0.000000000 1
2 -0.000000001 0.577350239 0.000000000 1
8 251 10
F F
1 1 C 2S 1 0 1
1 1 0.4920425600
1 2 0.0000000000
1 3 0.0000000000
1 4 0.0000000000
1 5 0.0030914953
1 6 0.0000000035
1 7 0.0006870343
1 8 0.0000000217
1 9 0.0008474838
1 10 0.0000001068
2 1 0.4918940160
2 2 0.0000000001
2 3 0.0000867961
2 4 0.0000000266
2 5 0.0030785331
2 6 0.0000000001
2 7 0.0006861789
2 8 0.0000000010
2 9 0.0008435850
2 10 0.0000000231
...
` 8 251 10` represent to
- norbitals
- npkt
- nbnd
` 1 1 C 2S 1 0 1` represent
- orbital index
- atom index
- atom type
- orbital name (principle quantum number n)
- Not sure
- Azimuthal quantum number (0 for s, 1 for p, etc)
- Magnetic quantum number (for p, have px, py, pz)
Can check like this
For graphene, as example, one carbon atom have
- one 2s orbital
- three 2p orbitals
And total two carbon atoms, so it can be found that
1 1 C 2S2 1 C 2P3 1 C 2P4 1 C 2P5 2 C 2S6 2 C 2P7 2 C 2P8 2 C 2P
plotband.x
Similar to simple plotband.x, but the projected file makes some difference
Remember to go into ./PROJ/ if you created and stored _bands.dat and _bands.dat.proj into
> plotband.x
Input file
> _bands.dat
Reading 10 bands at 251 k-points
List of atomic wavefunctions:
> 1 5
Range: -23.7690 10.4000eV Emin, Emax, [firstk, lastk]
> -25,12
high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 0.0000
high-symmetry point: 0.3333 0.5774 0.0000 x coordinate 0.6667
high-symmetry point: 0.5000 0.2887 0.0000 x coordinate 1.0000
high-symmetry point: 0.0000 0.0000 0.0000 x coordinate 1.5774
output file (gnuplot/xmgr) > gnuplot
bands in gnuplot/xmgr format written to file:
> gnuplot
output file (ps)
> ps
Efermi
> -3.3674
deltaE, reference E (for tics)
> 5 -3.3674
bands in PostScript format written to file: ps
output file for projected band (gnuplot script)
> gnuplotscript
run "gnuplot gnuplotscript" to get "gnuplotscript_projected.ps"
and/or run "ps2pdf gnuplotscript_projected.ps" to get "gnuplotscript_projected.pdf"
Files not useful (can delete):
- gnuplot x.x
- ps
Additional setting compared to simple band structure:
-
List of atomic wavefunctions:- orbital index in _bands.dat.proj
- here picked the two 2S orbitals (1,5, can be found in _bands.dat.proj)
-
Efermi- Fermi energy from
nscf.out
- Fermi energy from
-
deltaE, reference E (for tics)- tics interval in plotting
- reference usually pick Fermi energy again
-
output file for projected band (gnuplot script)- the code will generate a gnuplot script for plotting, look like
#!gnuplot
set terminal postscript portrait enhanced color dashed lw 1 ",12"
set output "gnuplotscript_projected.ps"
set xrange [0.0: 1.5774]
unset xtics
#set yrange [ -25.000000: 12.000000]
set ytics -25.000000, 5.000000, 12.000000
set ylabel "E - E_{ref} (eV)"
set border lw 0.5
set style arrow 1 nohead front lw 0.5 lc rgb 'black'
set arrow from 0.0000,graph 0 to 0.0000,graph 1 as 1
set arrow from 0.6667,graph 0 to 0.6667,graph 1 as 1
set arrow from 1.0000,graph 0 to 1.0000,graph 1 as 1
set title 'gnuplotscript_projected' noenhanced
plot 'gnuplot' u 1:($2 - -3.367400):3 w l palette lw 1 notitle, \
0.000000 lt 2 lw 0.5 lc rgb 'grey50' notitle
Comment out the first line set terminal ...
then gnuplot -p gnuplotscript will show up a gnuplot window and a .ps file gnuplotscript_projected.ps
and then ps2pdf gnuplotscript_projected.ps will convert the .ps file to a pdf one
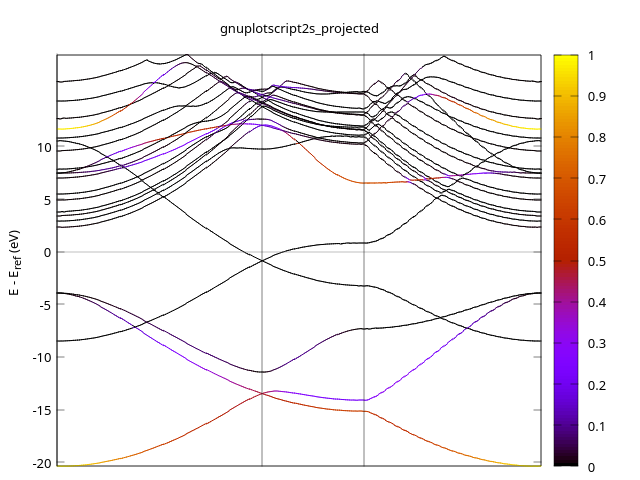
Can see the 2S orbitals contribute less (or even no contribution) near Fermi level
further analysis
can be done by inspect the contribution of others orbitals, by changing List of atomic wavefunctions: in plotband.x
can use the following codes for faster plotting (graphene case):
plotband.x input (2s for example)
_bands.dat
1 5
-25 12
gnuplot2s
ps
-3.3674
5 -3.3674
gnuplotscript2s
run.sh
#!/bin/sh
cd ./PROJ
element='C' # can be atom specific
for i in 2s 2p1 2p2 2p3;do
plotband.x < ${element}${i}.in > out.ppbands${element}${i}
sed -i '2s/^/#/' gnuplotscript${element}${i}
sed -i '3s/^/#/' gnuplotscript${element}${i}
sed -i '2s/$/\nset term png/' gnuplotscript${element}${i}
sed -i '3s/$/\nset output "'${element}${i}'.png"/' gnuplotscript${element}${i}
gnuplot -p gnuplotscript${element}${i}
done
rm gnuplot*.1
rm ps
- 2S
- 2P1
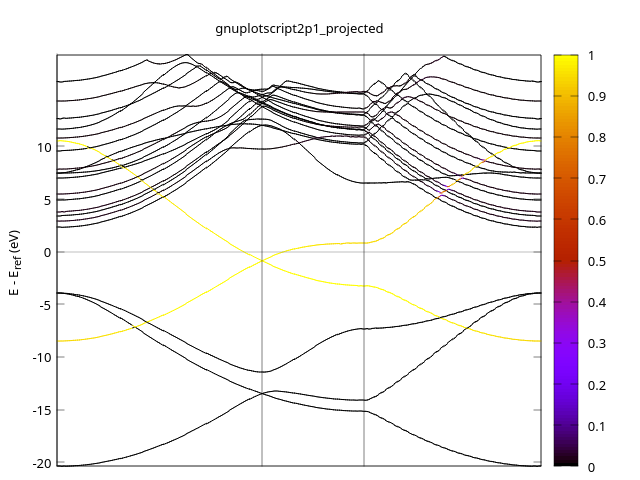
- 2P2
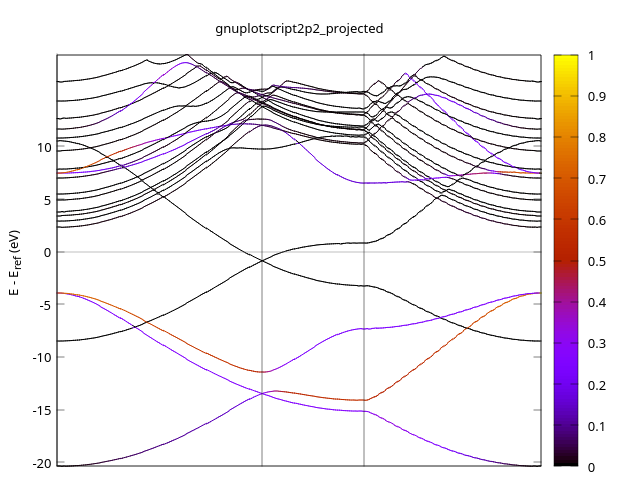
- 2P3
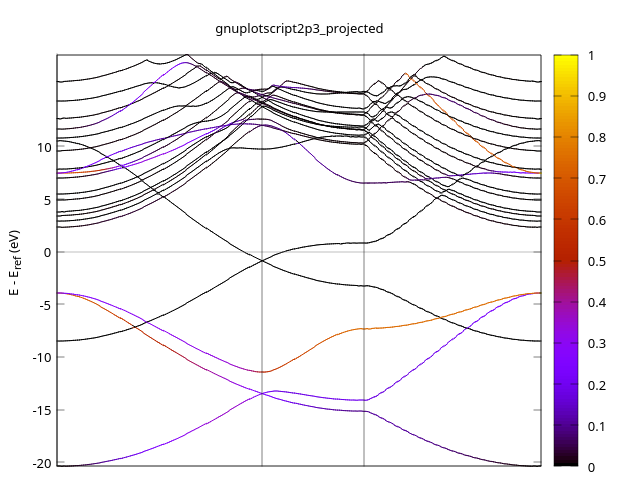
By this it is clear that the atomic orbital in graphene should be sp2 hybridization + pz (2p1 here), in which pz contribute the most to the states near Fermi level

Leave a Comment